

KaryoSolver is our proprietary solution for molecular karyotyping from low-pass whole genome sequencing (lpWGS), tested on Nanopore data. It leverages very fast/real-time sequencing to generate genome-wide copy number profiles, identifying large chromosomal abnormalities, microdeletions/duplications, and mosaicism.
Unlike traditional array-CGH, KaryoSolver provides:
– Scalable resolution in short time (hours, depending on sequencing time).
– Mosaic fraction estimation, enabling detection of clonal heterogeneity.
– Genome-wide coverage without probe design limitations, minimizing blind spots.
KaryoSolver module is part of 4eVAR platform, and builds on the academic framework developed by Prof Magi and his group in UNIFI.
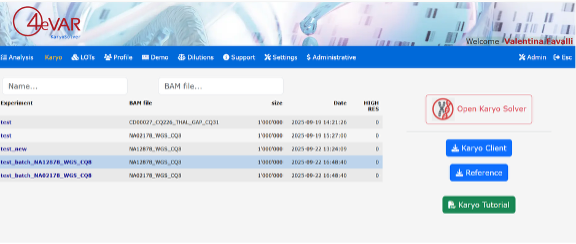
Figure 1: 4eVAR – KaryoSolver dashboard
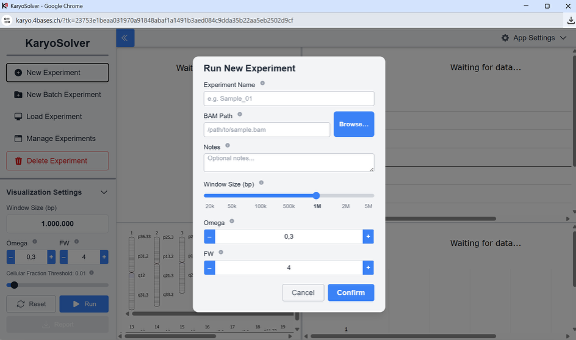
Figure 2: KaryoSolver modal to load BAM file.
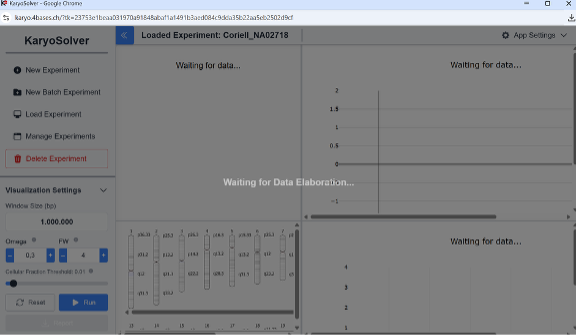
Figure 3: “Waiting for Data Elaboration” window.
In about a minute you will have genome-wide binning and segmentation with three main parameters:
• window_size: bin size (smaller = higher resolution, more noise).
• Omega (OM): segmentation sensitivity (higher = more sensitive, noisier).
• Filtered Windows (FW): minimum consecutive bins for a CNV call (higher = fewer noisy calls).
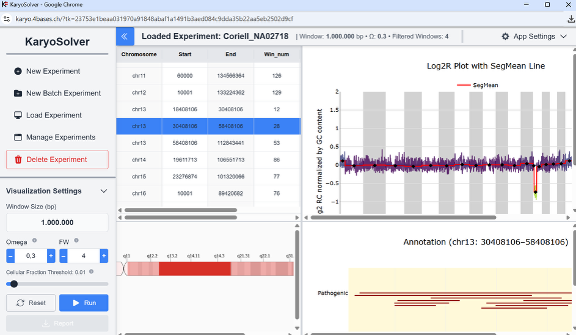
Figure 4: KaryoSolver dashboard.
Output: an interactive molecular karyotype profile, with tables and plots for exploration and annotation.
For best results, we recommend following the 4bases Technote for KaryoSolver sample preparation (RUO version):
This ensures consistent coverage profiles and reliable CNV detection.
KaryoSolver provides progressive resolution as sequencing time increases. This makes it directly comparable to Agilent array-CGH platforms, but with faster turnaround and flexible resolution.
Platform | Typical Resolution | Processing/Sequencing Time | Turnaround |
60K CGHarray | ~150 kb | 24–40 h hybridization + 6–8 h prep | 2–3 days |
400K CGHarray | ~30–50 kb | 24–40 h hybridization + 6–8 h prep | 2–3 days |
1M CGHarray | ~20–30 kb | 24–40 h hybridization + 6–8 h prep | 2–3 days |
KaryoSolver | 10Mb >2h 100kb>6h 30kb>24h | Fast sequencing | Same day (6–24 h)* |
* Standard ONT rapid or optimized multiplex-ready 4bases RDC3290 preparation kit with ligation library prep V14.
KaryoSolver allows interactive exploration of CNVs:
– A whole-genome plot displays normalized log2 ratios across all chromosomes for rapid visualization of alterations.
– Clicking on a chromosome segment in the plot highlights the CNV in detail with annotation.
– The Cellular Fraction Threshold can be adjusted to hide low-fraction events and focus on clinically relevant CNVs.
– Users can refine results by tuning OM and FW values iteratively to balance sensitivity against noise.
KaryoSolver is fully integrated with ClinGen resources for ACMG-compliant CNV interpretation. CNVs identified are annotated with overlapping known pathogenic regions, dosage-sensitive genes, and databases such as DECIPHER and ClinVar. This ensures that each finding is contextualized within clinically validated knowledge.
Normalization and log2 ratio calculation are at the core of KaryoSolver’s CNV calling:
– Each genomic window (bin) is normalized against expected read counts.
– Log2 ratios are computed as log2(observed/expected coverage).
– Typical thresholds:
• ~0.0 → diploid (2 copies).
• –0.5 → heterozygous deletion (1 copy).
• –1.0 → homozygous deletion (0 copies).
• +0.58 → single duplication (3 copies).
Mosaicism: refers to the presence of two or more genetically distinct cell populations derived from a single zygote within the same individual. In KaryoSolver, mosaicism is inferred when the log2 ratio deviation is intermediate (e.g., –0.25 for a 50% mosaic heterozygous deletion).
Reference (required)
Use GRCh38 no-alt analysis set (UCSC IDs) to align with ACMG guidance for large CNVs.
Files: the FASTA (…no_alt_analysis_set.fasta or .fna) and its index (.fasta.fai or .fna.fai).
You may also use the bundled copies shipped with the local client in /data folder:
/data/references/hg38/GCA_000001405.15_GRCh38_no_alt_analysis_set.fasta
/data/references/hg38/GCA_000001405.15_GRCh38_no_alt_analysis_set.fasta.fai
Alignment (concepts)
Why this matters
Correct reference choice and sorted/indexed BAMs are essential for robust read-counting, segmentation, and ACMG-consistent large-CNV interpretation.